I recently stumbled upon a very interesting question on one of RDKit's forum, asking about the tracking of atoms through chemical reactions, and this really resonated with me. The reason behind it not only being that I struggled with it many times before being able to find some reliable go-to methods, but also that it was not the first time I heard that question. With that little blog post, I hope to share with you some methods that you will find useful and that you will be able to implement very easily in your workflows.
2. Aligning two similar structures
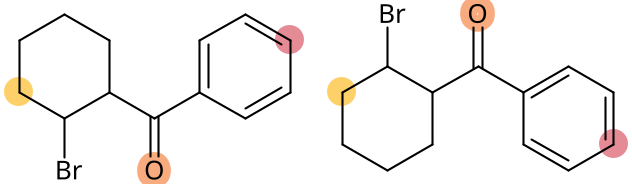
Two similar molecules with aligned highlighted atoms
The first thing I would like to tackle with this post is the task of aligning atom indices of two similar molecules, which is a task less trivial than it seems. Taking RDKit as an example, the indices of atoms are not invariant, and the order in which the atoms are initiated depends on the SMILES, given one creates them with their function Chem.MolFromSmiles(). For this task, my go-to method is now to always use the canonicalRank of the atoms. To make my point clearer, an example is shown below:
from rdkit.Chem import MolFromSmiles, MolToSmiles, CanonicalRankAtoms
# Let's instantiate a molecule, an equivalent but unaligned molecule
# and let's assume we know the index we are interested in from some previous code
idx_of_interest = 12
my_smiles = "c1ccccc1C(=O)C2CCCCC2Br"
my_mol = MolFromSmiles(my_smiles)
my_alt_smiles = "BrC1CCCCC1C(=O)c1ccccc1"
my_alt_mol = MolFromSmiles(my_alt_smiles)
# You can artificially create some different ones with: MolToSmiles(my_mol, rootedAtAtom=n) with n!=0
# We can ensure they are the same molecule by comparing their canonical SMILES
# N.B. Make sure your mols are unmapped to do that simplistic test below as the
# canonical SMILES can be affected by mapped atoms
assert MolToSmiles(my_mol) == MolToSmiles(my_alt_mol),(
f"Your molecules are not the same, smiles are {MolToSmiles(my_mol)} and {MolToSmiles(my_alt_mol)}"
)
# And we can also check that they are not aligned (Please note that it is not yet a formal test, I just look at symbols)
assert [a.GetSymbol() for a in my_mol.GetAtoms()] != [a.GetSymbol() for a in my_alt_mol.GetAtoms()],(
f"Your symbols are all aligned"
)
print("All assertions passed")
# Here, the cleanest methods I found is to have a dictionary of original_idx: alt_idx
# using the canonical rank (breakTies=True is very important here)
canrank = [r for r in CanonicalRankAtoms(my_mol, breakTies=True)]
alt_canrank = [r for r in CanonicalRankAtoms(my_alt_mol, breakTies=True)]
#print(canrank)
#print(alt_canrank)
conversion_dict = {
i: alt_canrank.index(canrank[i]) for i in range(len(canrank))
}
#print(conversion_dict)
alt_idx_of_interest = conversion_dict[idx_of_interest]
print(f"The index of original idx {idx_of_interest} in the alternative mol is {alt_idx_of_interest}")
print(f"Canonical rank is {canrank[idx_of_interest]}")
# Finally, we could just check that this result is correct by tagging the atoms in both mol objects
my_mol.GetAtomWithIdx(idx_of_interest).SetAtomMapNum(1)
my_alt_mol.GetAtomWithIdx(alt_idx_of_interest).SetAtomMapNum(1)
print(MolToSmiles(my_mol))
print(MolToSmiles(my_alt_mol))
assert MolToSmiles(my_mol) == MolToSmiles(my_alt_mol),(
f"Your molecules are not the same, smiles are {MolToSmiles(my_mol)} and {MolToSmiles(my_alt_mol)}"
)
3. Keeping track of atoms during a known transformation
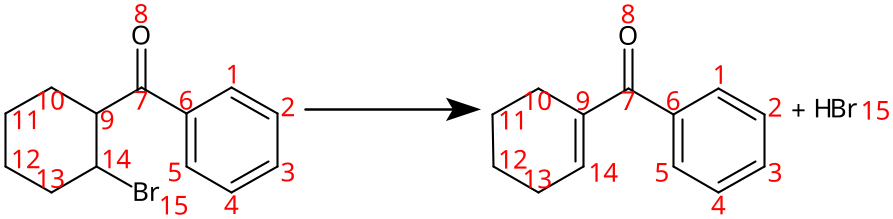
Reaction with atoms tracked
Even though methods like RXNMapper (Schwaller et al., 2021) or LocalMapper (Chen et al., 2024) can be very efficient tools of Atom-to-Atom Mapping (AAM) when the transformation is unknown, the use of ML techniques is not necessary in the precise case of a known transformation. In this paragraph, I want to showcase a method to track atoms when transformations are based on templates, a tool used very broadly to describe a chemical transformation (Coley et al., 2019; Genheden et al., 2020; Chen et al., 2024, and much more...). This task is more complex than the previous one, and one of my go-to methods for keeping track of atoms is the isotopic labeling. Once again, this small snippet should help you understand very easily how this trick works.
from rdkit.Chem import MolFromSmiles, MolToSmiles, rdChemReactions
# For this example let's say I want to apply an E2 elimination to my molecule
# First I will get the E2 elimination SMARTS, in this case, I wrote this one by hand
# But some useful packages can help you construct them, for example rxnutils
# (https://molecularai.github.io/reaction_utils/rxnutils.chem.html)
temp_sma = "[C;!H0:1][C:2][Br:3]>>[C:1]=[C:2].[BrH:3]"
template = rdChemReactions.ReactionFromSmarts(temp_sma)
# And I want to apply that template on my molecule of interest. Let's also
# assume that you know the index you are interested in from some previous code
my_reac_smi = "c1ccccc1C(=O)C2CCCCC2Br"
my_reac_mol = MolFromSmiles(my_reac_smi)
for idx, a in enumerate(my_reac_mol.GetAtoms()):
# Setting AtomMap idx or custom property doesn't work, and will be erased by the
# RunReactants method. The only property that is not erased seems to be the isotope,
# Hence, let's do some isotopic labeling
a.SetIsotope(idx + 1) # Careful to +1, because isotope 0 means no info on the isotope
# Feel free to try these to understand at which point we lose them
#a.SetIntProp('tag', idx+1)
#a.SetAtomMapNum(idx+1) # Careful to +1, because atom map 0 means no mapping
prods_tuple = template.RunReactants((my_reac_mol,))
assert len(prods_tuple)>0, "Template could not be applied on that reactant set"
for prod in prods_tuple[0]:
for a in prod.GetAtoms():
a.SetAtomMapNum(a.GetIsotope())
a.SetIsotope(0)
for a in my_reac_mol.GetAtoms():
a.SetAtomMapNum(a.GetIsotope())
a.SetIsotope(0)
map_prod = '.'.join([MolToSmiles(p) for p in prods_list])
map_reac = MolToSmiles(my_reac_mol)
print(f"{map_reac}>>{map_prod}")
4. Application in visualization and green chemistry
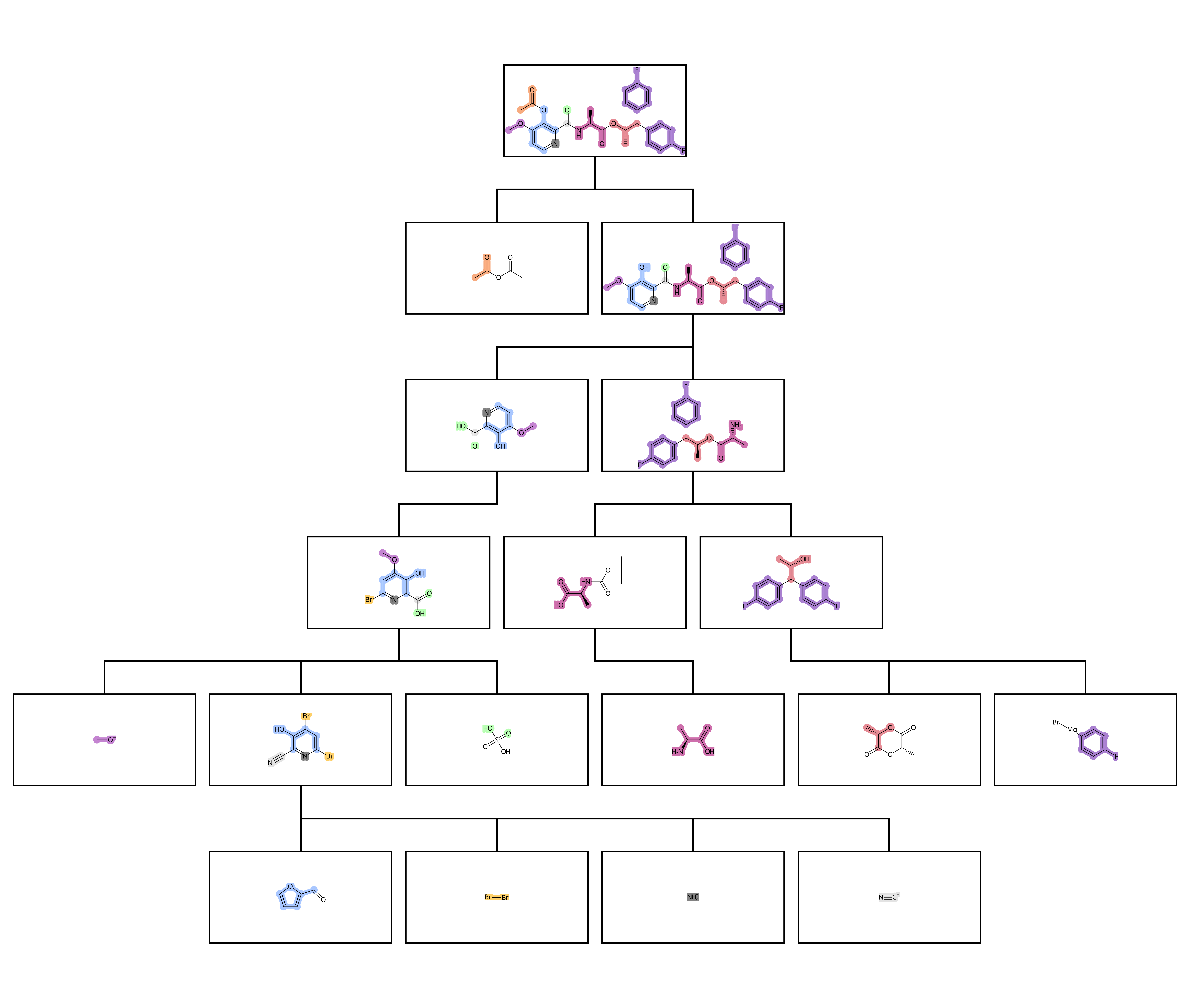
Now that you know how to track your atoms, I'd like to share with you an example application that can be unlocked by a powerful tracking of atoms. For this example, I got inspired by the synthesis of Florylpicoxamid (Babij et al., 2020). Because we now know how to map a reaction, we could map every single step of a known multistep synthesis. Then, it would be fairly easy to detect the species appearing among the reactants of a reaction, and among the products of another one. Once these species are detected, we could align their mapping in all reactions they are involved, using the "similar structure alignment". Finally, we can choose one color per starting material and propagate these colors in the whole tree, giving the very pleasing tree you can see below. A very concrete application of such a script could also be to track all atoms of a multistep synthesis to automate the calculation of the atom economy, which is one of the 12 principles of green chemistry.
Aligned mapping on the synthesis tree of Florylpicoxamid
5. Conclusion
With these two little snippets, you have two different techniques to align both similar molecules, and molecules that undergo known transformations, which for me are the two most important aspects to never lose track of any of your atoms. And I hope to have motivated why it could be interesting with the application above.
As it is the first blog post I wrote on this website, I hope you enjoyed the read, and if you want to exchange further on this or another topic touching chemistry on computers, don't hesitate to contact me or connect with me on my different profiles.